Reaction Simulation
The simulation of chemical structures and their reactivity has become so efficient and powerful that it percolates the organic chemistry literature nowadays. However, in many instances it is either used as an afterthought to rationalize experimental observations or to complement experimental mechanistic evidence. In our group, we use reaction simulations as a guide to molecular design. By simulating catalyst performance for a wide range of substrates and combining these results with experimental observations, we reduce the overall number of experiments that are necessary and accelerate catalyst optimization.
Automated Reactivity Exploration
State of the art simulation of chemical reaction mechanisms largely relies on a significant amount of trial and error. The high dimensionality of the associated space together with the computational expense of sufficiently reliable approaches makes a comprehensive exploration of all conceivable pathways intractable with currently available resources. Nevertheless, the use of efficient hybrid models relying on a combination of quantum mechanics and molecular mechanics models together with enhanced sampling techniques such as metadynamics allows for sufficient computational investigation of a pre-defined subset of target pathways. We implement these approaches in our closed-loop catalyst and reaction optimization workflow and develop new efficient and modular approaches allowing for more robust automated protocols for the simulation of reactivity.
Selected Publications
June 4, 2022
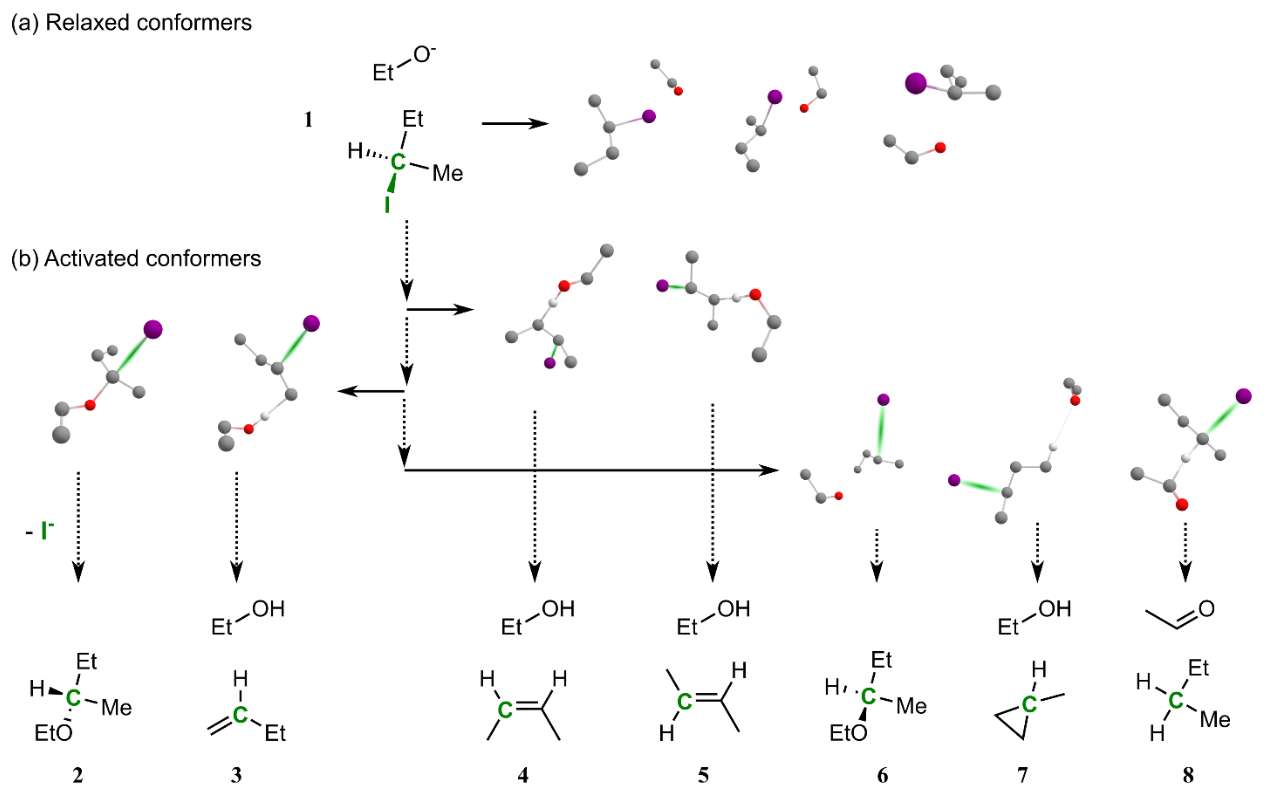
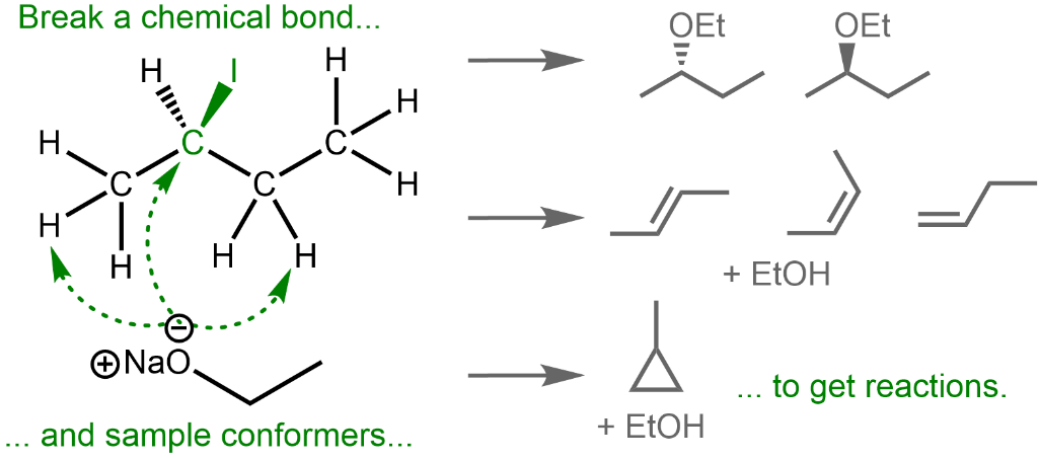
R. Rahrt, B. Hein-Janke, K. N. Amarasinghe, M. Shafique, M. Feldt, L. Guo, J. N. Harvey, R. Pollice, K. Koszinowski*, R. A. Mata*
J. Phys. Chem. A 2024, 128, 4663.
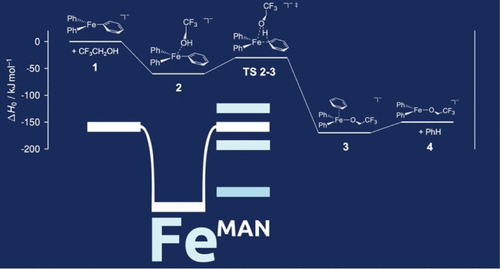
January 31, 2024
R. Pollice*, B. Ding, A. Aspuru-Guzik*
Matter 2024, 7, 1161.
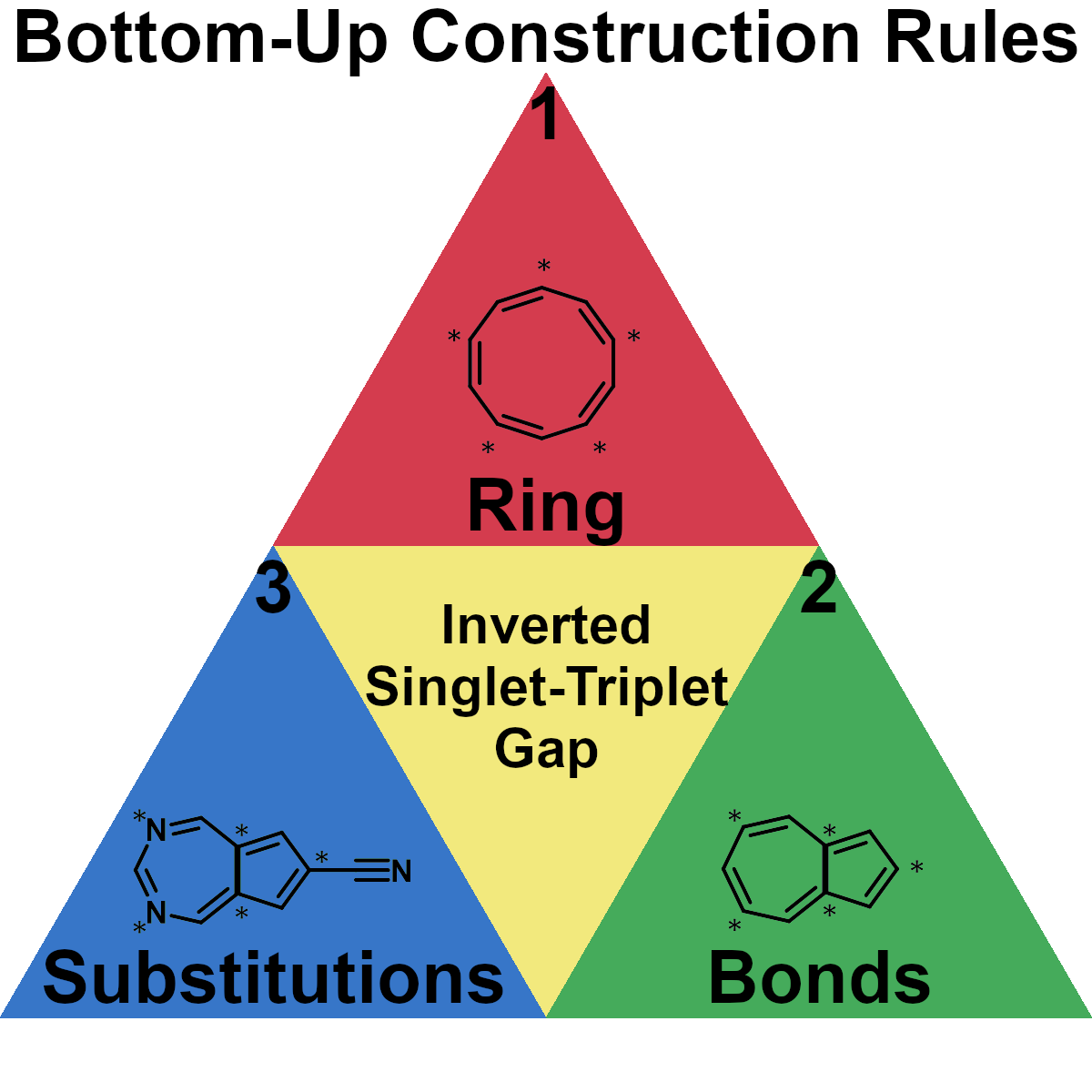
November 10, 2022
C. Lavigne, G. d. P. Gomes#,*, R. Pollice#,*, A. Aspuru-Guzik*
Chem. Sci. 2022, 13, 13857.